How to use LAMMPS on Seawulf
LAMMPS stands for Large-scale Atomic/Molecular Massively Parallel Simulator. It's a classical molecular dynamics (MD) code. As the name implies, it's designed to run well on parallel machines, but it also runs fine on single-processor desktop machines. http://lammps.sandia.gov/index.html
Current Version
There are currently three versions of LAMMPS available on Seawulf, gcc Intel builds, and a GPU-optimized gcc build.
The MPI enabled builds are LAMMPS versions 23Mar2023 and 2Aug2023, and include the following packages:
| BODY | DIPOLE | EXTRA-PAIR | MANYBODY | MOLECULE | POEMS | SHOCK |
| COLLOID | DRUDE | GRANULAR | MC | OPT | REAXFF | SRD |
| COMPRESS | EXTRA-DUMP | KIM | MEAM | ORIENT | REPLICA | |
| CORESHELL | EXTRA-MOLECULE | KSPACE | MISC | PERI | RIGID |
The GPU optimized build are LAMMPS versions 3Mar20 and 2Aug2023 and includes all of the previous packages with the addition of open-mp, gpu, and kokkos.
For more information these packages see: https://docs.lammps.org/Packages_list.html
The table below displays the latest version of LAMMPS and the associated hardware available for each version:
| HARDWARE | MODULE VERSION(S) |
| AMD Milan 96-core nodes | lammps/intel/2023.1/2Aug2023 |
| Intel Skylake 40-core nodes | lammps/intel/2023.1/2Aug2023 |
| Intel Sapphire Rapids 96-core nodes | lammps/intel/2023.1/2Aug2023 |
| Nvidia A100 gpu nodes | lammps/a100-gpu/2Aug2023 |
| All other GPU Nodes including P100 and V100 | lammps/gpu/3Mar20 |
| Intel Haswell 28-core nodes | lammps/intel/2022.2/23Mar2023 |
Using LAMMPS
The current version of LAMMPS is available using both the Intel and GNU toolchains. Both versions are accessible through modulefiles; lammps/gcc12.1/mvapich2/03Aug2022 or lammps/intel/2022.1/2Aug2023
As usual, we recommend the use of the Intel compiled versions unless your workflow demands otherwise.
To access LAMMPS load the appropriate modulefile:
module load lammps/intel/2022.2/03Aug2022
or
module load lammps/gcc12.1/mvapich2/03Aug2022
Loading either of these modules also loads the appropriate implementation of MPI
Using GPU Accelerated LAMMPS
GPU accelerated lammps is only available using the GNU toolchain. This can be accessed through the lammps/gpu/11Aug17 module.
module load lammps/gpu/11Aug17
Open-mp threading can further be requested through setting the OMP_NUM_THREADS variable to the desired number of threads, for example 2:
export OMP_NUM_THREADS=2
LAMMPS Examples
Parallel LAMMPS
Once you have loaded one of these modules, the lammps binary will be available in your path as either: lmp_mvapich2 or lmp_intel
An example job script and basic input files are available in the LAMMPS_EXAMPLES directory.
The existing script files in the lammps_example directory are .pbs files, so you may need to convert the Torque instructions from #PBS to #SBATCH for Slurm. To test this workflow:
module load slurm module load lammps/intel/2022.2/03Aug2022 cd $LAMMPS_EXAMPLES mkdir -p $HOME/lammps_example cp * $HOME/lammps_example && cd $_ sbatch lammps_intel.slurm
This should produce the following files:
dump.melt.gz, log.lammps, and my_test.o*
LAMMPS GPU
The following script can be used as a template for LAMMPS GPU submission:
#!/bin/bash #SBATCH --nodes=1 #SBATCH --ntasks-per-node=28 #SBATCH --time=01:00:00 #SBATCH --job-name=my_test #SBATCH -p gpu mkdir $HOME/lammps/gpu_test cd $_ module load mvapich2/gcc/64/2.2rc1 module load lammps/gpu/3Mar20 cp $LAMMPS_EXAMPLES/in.melt . export OMP_NUM_THREADS=2 export MV2_ENABLE_AFFINITY=0 mpirun -np 28 lmp_gpu -sf gpu -pk gpu 8 < in.melt 2> out.txt 1> err.txt
The variables OMP_NUM_THREADS and -pk gpu # can be varied to suit your needs.
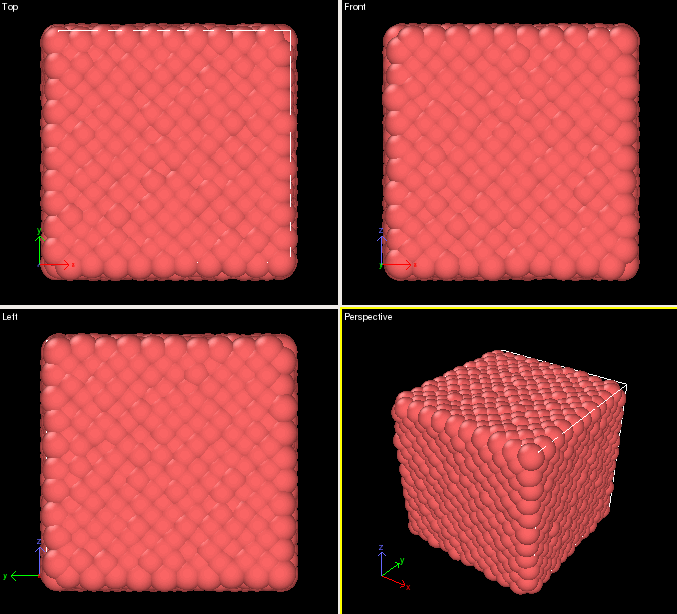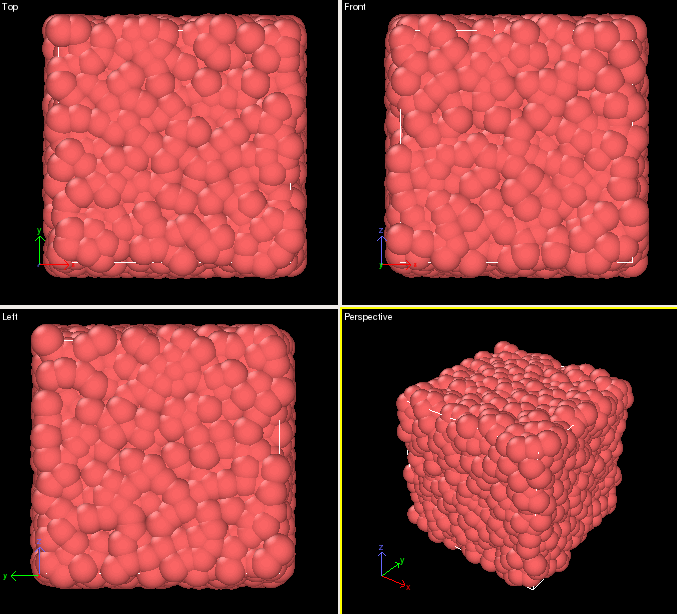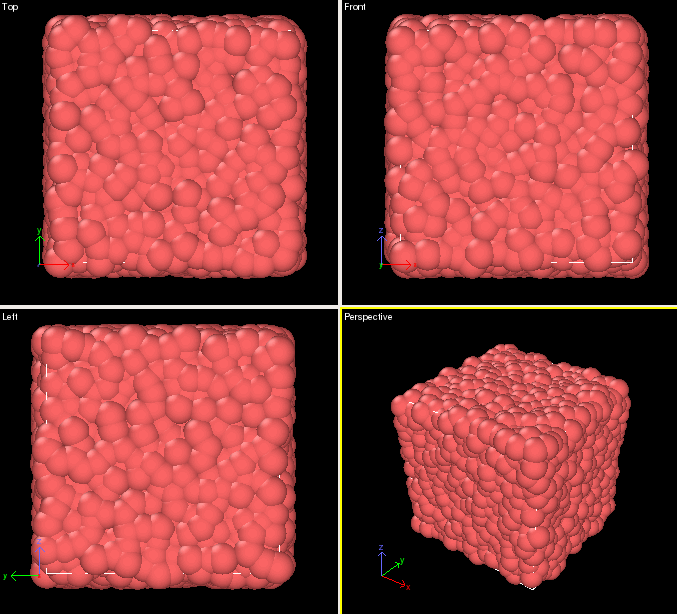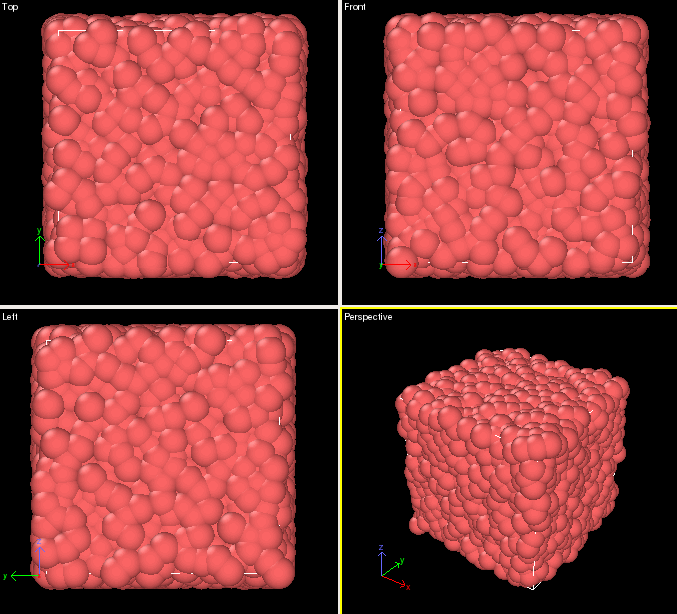
Visualizing the Results
Several tools exist for visualizing the results of the above calculation. However, for quick visualizations on the cluster, we recommend an application such as OVITO: https://ovito.org/
The output file from the above calculation, dump.melt.gz, can be visualized by first loading the ovito module, then calling ovito:
module load ovito/2.8.2 ovito dump.melt.gz
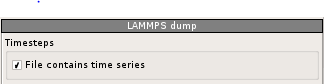
To view successive iterations as an animation check the "File contains time series" box
Which should produce the following:
For More Information Contact
Still Need Help? The best way to report your issue or make a request is by submitting a ticket.
Request Access or Report an Issue